hpRNA
Hamiltonian paths for virus genome organization
This project is maintained by hpRNA
hpRNA
v. 15/01/2015
James Geraets, University of York
jg923@york.ac.uk
In many single-stranded RNA viruses, multiple contacts between viral genome and capsid protein (packaging signals, PSs) play functional roles during virus assembly. The details of these contacts, in terms of the positions, connectivity and level of occupancy of the potential binding sites, place structural constraints on the organization of the viral genome within the capsid. Depending on the PS-mediated assembly mechanism adopted, and the specific roles of the PSs, these result in different types of constraints. One type of constraint, which applies to particles in which the majority of the PS binding sites are occupied and for which an icosahedrally-averaged cryo-EM map reveals the packaged genome in proximity to capsid as a polyhedral RNA cage, can be formulated in terms of Hamiltonian paths. The Hamiltonian path constraints depend on the topology of the RNA cage.
We provide here the code for the computation of such Hamiltonian path constraints for the polyhedral cage corresponding to bacteriophage MS2, and we comment on how the code could be generalized to accommodate RNA cages with other topologies. In a related publication [forthcoming in 2015] we demonstrate how the library of Hamiltonian path constraints can be used to interrogate an asymmetric cryo-electron tomographic reconstruction of MS2 to reveal a highly constrained conformation of the packaged genome.
GLP v3 license applies: see separate file for information.
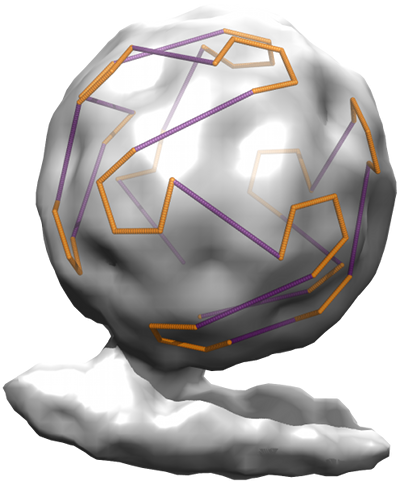
The figure shows a Hamiltonian path constraint superimposed on a tomogram of bacteriophage MS2.
CONTENTS
- Introduction
- Requirements
- Guide
- Configuration
- License
- Maintainance
INTRODUCTION
This code, as supplied, generates Hamiltonian paths as models for the RNA organization in proximity to capsid for ssRNA viruses, initialized for the bacteriophage MS2. A script is also supplied that allows the user to apply constraints to the library of Hamiltonian paths, thus enabling the addition of constraints arising from further structural insights, such as restriction to circular Hamiltonian paths, that narrow down the possible path solutions. The software is provided "as is", but the author is willing to correspond to help anyone with interest to amend/adapt the code, to interrogate asymmetric structures of other viruses and determine possible RNA conformations. A link to a scientific manuscript describing the process will be added here upon publication.
REQUIREMENTS
The software is coded in python 2.7, a scripting language that is very easy to use and adapt. More information on python can be found at python.org or greenteapress.com/thinkpython/
Required python modules are:
Other required software:
GUIDE
Step-by-step guide to recreating results for MS2.
- Generating Hamiltonian paths
Using hpRNA_generate.py the user can generate Hamiltonian paths that relate to the geometry of the RNA density in proximity to capsid. In the case of bacteriophage MS2, the geometric cages is as shown in Fig. 1b in the manuscript, with edges connecting the 60 packaging signals (PSs) in contact with capsid protein. This geometry is specified within the script. Therefore, the script as provided can be used for MS2, GA or any Leviviridae with RNA cages of the same topology (i.e. polyhedral cages with the same number of vertices and edge connections between them, irrespective of edge length or relative orientations of the edges).
However, the neighbour maps and general geometry in the script can easily be substituted to make the code applicable to any cage of interest, and thus treat viruses with different geometries. If that were desired, other alterations would also have to be made in following steps in the code. For example, the number indicating the length of the path would have to be adjusted (note that for MS2, all paths are between 60 positions in the cage).
We have utilised several steps to speed up the derivation of the Hamiltonian paths. However, for clarity, many of these have been rolled back for this open-source code. This may mean that some calculations using the script will take a very long time. Yet many special cases for MS2 have been coded (c.f. example_4 and example_6), and must be considered when adapting the code: for example, in MS2 the paths are effectively circular, because both 5' and 3' end bind to MP. In the code, this is specified as a vertex that the path can only visit at the start and end. This constraint will also apply to many other viruses, but should be omitted if there is no evidence of such circularization.
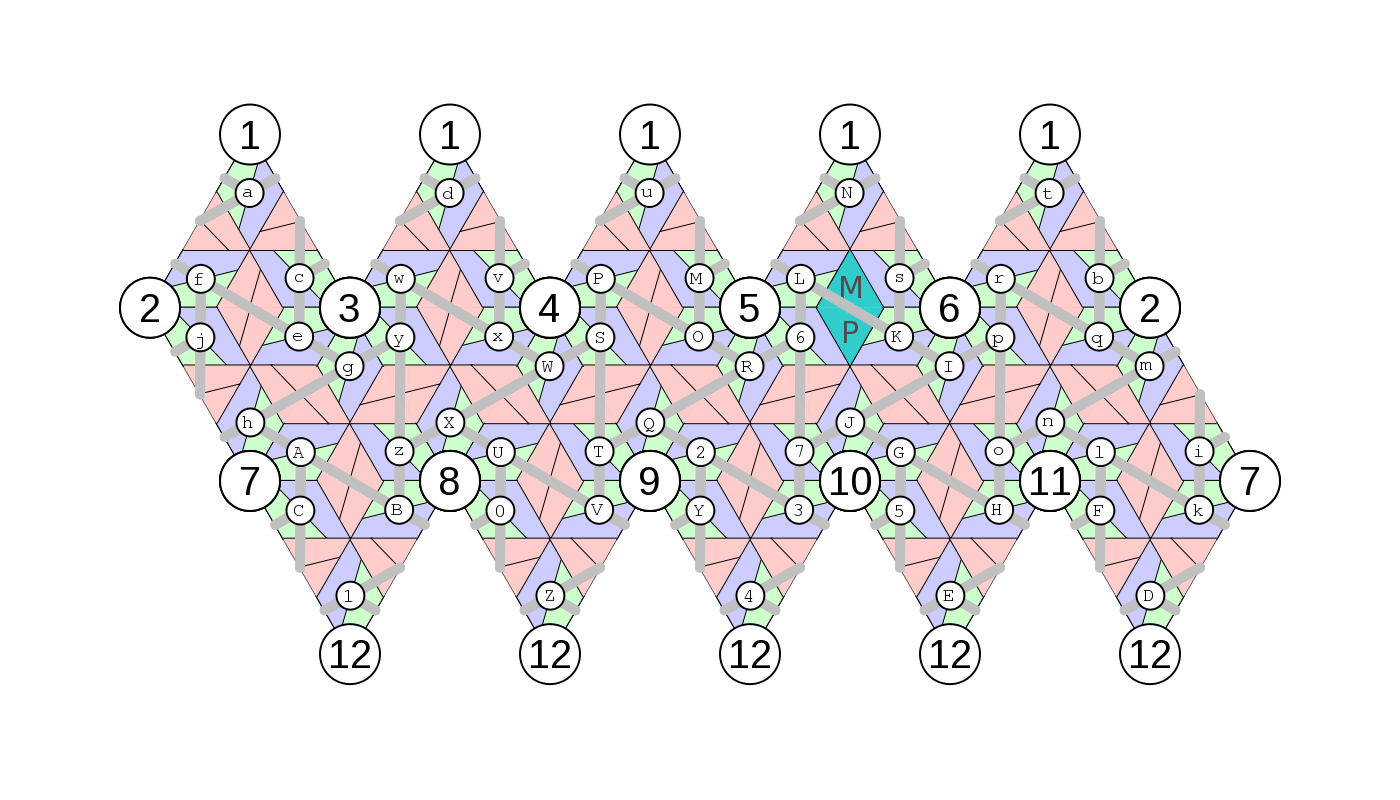
Paths are calculated with reference to the structural organisation of the capsid. For this, a labelling system is introduced, in which proteins are labeled as (cf "geometry_guide.png"): abcdefghijklmnopqrstuvwxyzABCDEFGHIJKLMNOPQRSUVTWXYZ01234567.
In the analysis presented in this paper, we ignore density associated with "short" edges of the polyhedron, i.e. only consider density related to long edges. That could be modified if higher resolution data were available for the short edges as well.
Once paths have been generated on the protein neighbour map, they can be generalized into moves, i.e. described as the order of mapping operations between proteins, rather than the proteins themselves. This allows an easier way to recreate the paths starting from any given protein. Also, calculation of symmetric and mirror paths is much easier when the protein positions are disregarded: these degenerate paths can be calculated with simple string manipulations.
- Mapping Hamiltonian paths onto the virus
Given the calculated generalized paths, we have to superimpose these paths onto the RNA density, starting and finishing at defined points with reference to the viral capsid. These starting points come from consideration of the binding of the RNA 5' and 3' end regions to the maturation protein, which localizes the ends of the RNA in the vicinity of MP. We have called this "realizing" individual paths, and this is undertaken in the script hpRNA_constrain.py. Starting points are referred to by the same protein labelling map utilised in the first step, see above.
- Difference maps
The analysis is based on a asymmetric structure of MS2, obtained via averaging of individual particles [Dent et al., Structure 2013]. The asymmetric structure was obtained by imaging mature MS2 bound to its natural receptor, the F-pilus of E. coli. The data can be found in the Electron Microscopy DataBank, with tag EMD-2365.
The X-ray structure of the protein capsid of MS2 can be found at the Protein Data Bank, with PDBID 2MS2. This protein structure was filtered to 39AA resolution to match the EM data. Then the pixel size and orientation of the two maps are made equivalent by trilinear interpolation of the reduced-resolution X-ray structure. A contour mask of 0.5sigma is used to sample the low-resolution map and used to eliminate the protein density. A similar difference map is created between an icosahedrally-averaged map [Toropova et al., J Mol Biol 2008] and the resampled filtered protein, yielding a symmetric cage of RNA with a polyhedral shape.
For both maps, the outer shell of RNA in proximity to capsid is isolated by icosahedral masking with vertex radii of 80AA and 120AA.
- Mapping data onto the geometric model
The polyhedral density was partitioned into segments attributed to the edges of the polyhedral cage of RNA seen in the symmetric map. Pixels from the asymmetric RNA map can be associated with defined segments on the polyhedral shell, and each connection thus has a density profile associated with it.
Some segments were not used further in the analysis if they had not sampled many pixels, or were in parts of the tomogram where other features would obscure the RNA density. In particular, connections adjacent to the MP/pilus are discarded as they may contain unmasked MP density.
- The density profiles
The densities of the connections were compared, and statements about the likelihood of RNA occupancy were made about these connections. These were noted as constraints.
The connections were named with reference to the vertices they connect. In particular, we labelled the vertices with reference to the capsid proteins, using again the labelling system introduced earlier, i.e. PS positions being labelled as proteins with the following letters and numbers: abcdefghijklmnopqrstuvwxyzABCDEFGHIJKLMNOPQRSUVTWXYZ01234567
"geometry_guide.png" shows the relation between the heterodimers and vertices. The maturation protein has been (arbitrarily and without loss of generality) chosen to map to the homodimer on the two-fold axis between vertices 5 and 6. This is marked on the path outputs.
- Constraining paths
Constraints were saved in a file, see constrain.txt as an example. The first two columns represent the identifiers of the proteins the edge is connecting. The next entry, a 0 or a 1, sets the edge either to 'unoccupied' or 'occupied'. E.g. a row in the constraints file reading ef 1 means that the edge between the positions 'e' and 'f' is constrained to be occupied in the analysis. Similarly, TQ 0 means that the between positions 'Q' and 'T' is constrained to be non-occupied in the analysis. Note that in the analysis carried out here, we were unable to make use of any data regarding short edges, as the resolution of the asymmetric tomogram was too low. However, information regarding long edges turned out to be sufficient for the analysis. All constraints are entered via the same mechanism.
- Drawing Hamiltonian path solutions
Figures of the Hamiltonian path solutions for ms2 can be displayed in 2d using hpRNA_ms2_draw.py. This could be amended to display solutions for other viruses. The special code for ms2 can be triggered by specifying the --ms2 tag in the hpRNA_constrain.py script.
CONFIGURATION
Given the above guide, and comments in the code, the program can be amended/adjusted to calculate Hamiltonian paths for a wide variety of scenarios, and additional constraints can be included if desired by the user. For support/collaboration, please contact jg923@york.ac.uk
LICENSE
License applies to software. Please see separate license file.
MAINTAINANCE
This software is currently maintained by James Geraets, University of York. Support is envisaged for at least a couple of years. Comments and queries should be addressed by email or to:
James Geraets
RCH\321 YCCSA
University of York
York
YO10 5GE
United Kingdom